罕見神經疾病Kufor-Rakeb綜合征、智力發育障礙等介紹及相關小鼠模型
談及神經精神疾病,人們腦海中往往浮現的是阿爾茲海默癥(AD)、帕金森病(PD)和抑郁癥這“三大巨頭”。不過,除了這些廣為人知的疾病之外,還有一些隱匿不露的“魔鬼”在默默影響著不少人的生活。這些疾病或源于相同的致病基因,或根據不同的致病基因表現出相似的疾病表型,鑒于疾病機制與癥狀的相似性,常常被誤診為常見的神經疾病,從而導致了不正確的研究和治療方案。因此,深入了解這些罕見的神經疾病顯得尤為重要。
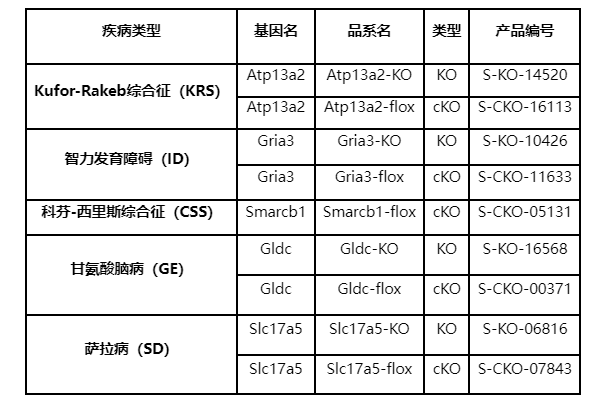
Kufor-Rakeb 綜合征(KRS)
Kufor-Rakeb綜合征是一種罕見的青少年起病的帕金森綜合癥(PD),屬于神經系統變性疾病。該綜合征的臨床癥狀包括運動障礙、強直性帕金森綜合癥、核上性眼肌麻痹、癡呆以及進行性彌漫性腦萎縮等,其病因與ATP13A2基因密切相關。
KRS與ATP13A2基因密切相關,該基因編碼一種內溶酶體蛋白,在細胞內負責質子泵運輸并在腦組織中發揮重要作用,人類ATP13A2基因突變已被發現與多種神經變性疾病相關,其中最為突出的是帕金森病(PD)。自噬-溶酶體途徑的功能障礙是帕金森病(PD)發病機制的一個關鍵特征,ATP13A2基因的突變導致其編碼的質子泵功能異常,導致細胞內溶酶體功能紊亂,進而引起神經元死亡,從而誘發帕金森病[1]。近年來,研究人員通過開發ATP13A2基因敲除小鼠模型,其內溶酶體蛋白Atp13a2的缺失導致小鼠出現膠質增生、泛素化蛋白聚集、運動障礙和神經元的功能異常等神經系統疾病表型以及自噬受損、鐵代謝紊亂、細胞因子異常和免疫反應異常等代謝和免疫表型,這些模型成功模擬了早發性帕金森病的一些特征,為進一步研究該疾病提供了有價值的工具[2-3]。
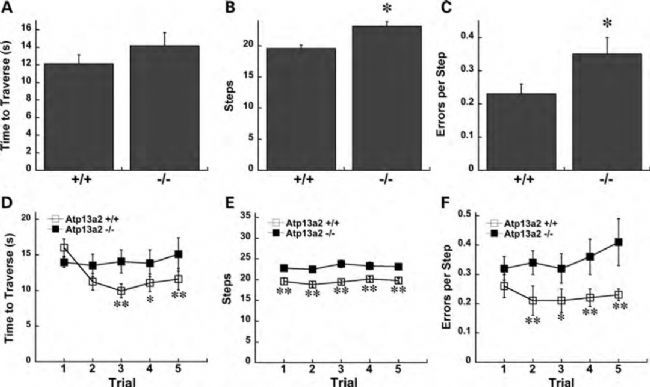
Atp13a2-KO小鼠出現運動障礙表型[3]
智力發育障礙(intellectual disability, ID)
智力發育障礙(Intellectual Disability, ID)是一種神經發育障礙,其主要特征為智力發育遲緩和認知能力受限。ID可能由遺傳因素、先天性疾病、感染和缺氧等多種原因引起。基因是智力發育的一個重要因素,已經證實許多基因與ID相關。除了常見的FMR1和MECP2,Gria3基因也是智力發育障礙的一個重要致病基因。Gria3編碼AMPA型谷氨酸受體(AMPAR)之一,在中樞神經系統中發揮著關鍵作用,通過調節神經元之間的信號傳遞、神經元的興奮性和突觸可塑性等方式來調節神經系統的功能。Gria3基因突變不僅與智力發育障礙有關,還可能導致多種神經系統疾病的發生,如自閉癥、帕金森病和阿爾茨海默病等[4]。
在小鼠中,敲除Gria3基因會導致海馬體中長期增強型突觸傳遞(LTP)的產生和維持方面存在缺陷,同時神經粘附分子也會異常,這會導致運動、協調性、認知和學習的障礙。此外,雄性小鼠還存在攻擊性增強的情況[5-6]。
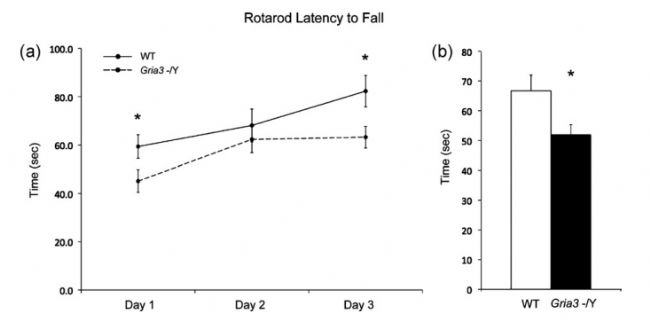
Gria3-KO小鼠動作協調性和平衡性存在缺陷
科芬-西里斯綜合征(CSS)
科芬-西里斯綜合征(Coffin-Siris syndrome, CSS)是一種先天性畸形綜合征,其特點是嚴重發育遲緩、生長障礙、肌張力低下、面部外觀明顯和特征粗大以及手腳趾發育不全等,這是由BAF復合體成分基因突變引起的疾病。研究表明,BAF復合體所有成分基因的突變皆與該疾病有關,其中SMARCB1的突變還與伴脈絡叢膜增生的智力障礙(ID-CPH)以及其他神經發育障礙有關[7-8]。
為了更好地理解SMARCB1基因突變的疾病機制,Filatova等人生成了神經干/祖細胞Smarcb1條件性敲除的小鼠,該小鼠表現出各種腦中線異常,與臨床上SMARCB1相關的CSS和ID-CPH患者的大腦缺陷相似[9]。這一研究結果有望幫助進一步探索SMARCB1的作用機制,并為治療CSS和ID-CPH等相關疾病提供新的思路和方向。
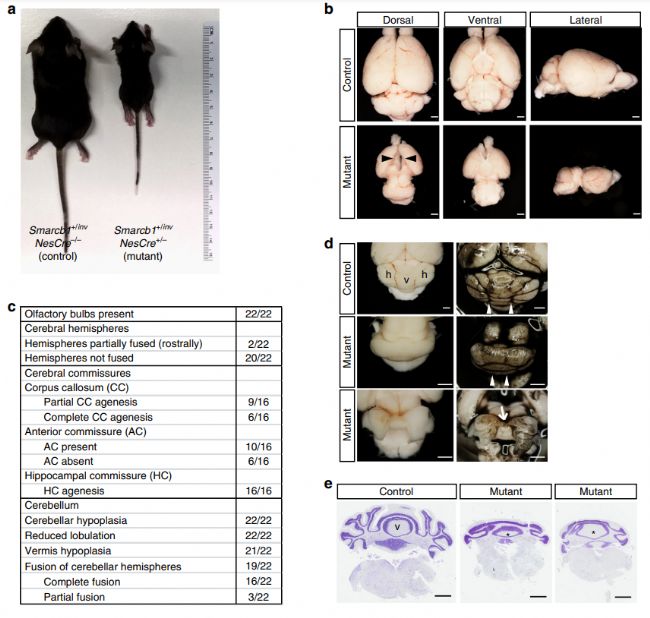
Smarcb1基因神經系統條件性敲除小鼠呈現嚴重的總體和腦部發育障礙[9]
甘氨酸腦病(GE)
甘氨酸腦病(Glycine Encephalopathy, GE),也稱非酮癥性高血糖癥(NKH),是一種罕見的代謝性疾病,對神經系統造成嚴重影響。該疾病由GLDC基因突變引起,導致甘氨酸脫羧酶功能異常。患者通常表現出智力發育障礙、痙攣和抽搐等癥狀,極度嚴重者甚至會導致死亡。GLDC基因編碼甘氨酸脫羧酶,參與甲基化代謝途徑中的甘氨酸代謝,甲基化代謝途徑在體內扮演著關鍵的角色,包括生成新的甲基基團、參與核苷酸代謝以及神經遞質代謝等。由于GLDC基因突變與GE的發生率高度相關,因此常被用于GE的診斷標志[10]。
GLDC致病突變會導致嚴重的神經系統病變,包括智力障礙、抽搐、肌張力紊亂等癥狀,小鼠實驗表明GLDC基因突變會影響小鼠的生長、行為和神經系統功能。敲除GLDC基因的小鼠會出現甘氨酸蓄積和神經元損傷,表現出與人類甘氨酸腦病相似的癥狀。這種小鼠模型被廣泛應用于GE發病機制研究、治療方法開發以及新型藥物的篩選,為深入研究該疾病提供了有價值的工具[11-12]。
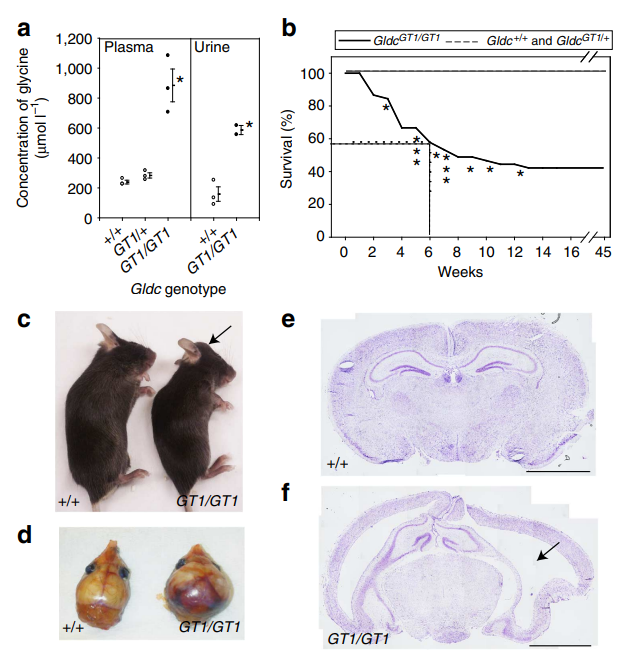
Gldc缺失小鼠(GT1/GT1)甘氨酸水平顯著提升且存在腦積水和腦室增大的表型[11]
薩拉病(SD)
薩拉病(Salla disease, SD)是一種常染色體隱性遺傳病,由SLC17A5基因突變引起,患者在臨床上表現為神經障礙和腦白質病變,隨著時間的推移病情逐漸惡化,最終導致智力退化、運動和語言障礙等癥狀。此外,SLC17A5基因的突變與某些類型的谷胱甘肽合成酶缺乏癥(GSSD)和遺傳性神經性減少性疾病(ISSD)有關。GSSD患者主要表現為溶血性貧血、代謝性酸中毒、焦谷氨酸尿癥和進行性神經系統疾病癥狀等,ISSD患者則表現為運動發育遲緩、震顫和抽搐等神經系統癥狀[13-14]。
在臨床前的研究中,通過敲除小鼠的Slc17a5基因可以模擬薩拉病的癥狀。這些小鼠表現出腦白質病變,類似于薩拉病患者的癥狀,同時還伴有智力障礙、共濟失調和肌張力不足等癥狀[13-15]。
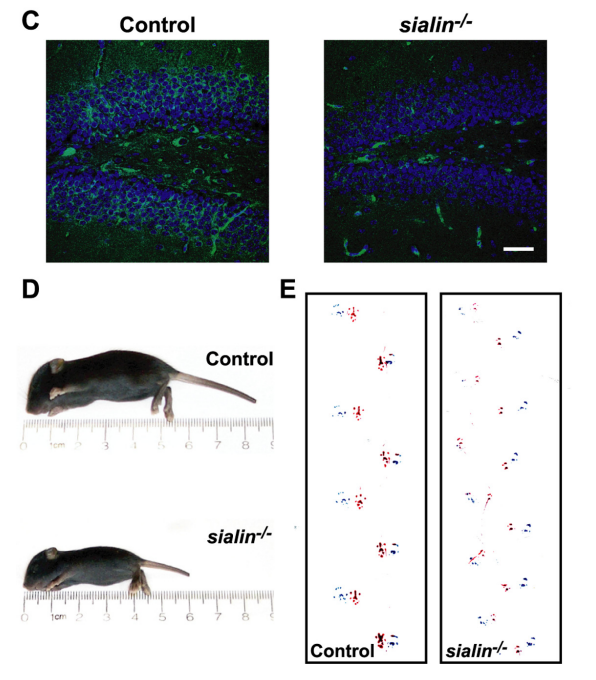
SLC17A5-KO小鼠(sialin-/-)表現為體型較小、發育不全、嚴重震顫和步態不協調表型[13]
賽業生物罕見病研究資源
基因編輯小鼠
小鼠基因編輯模型在罕見病機制研究和藥物研發評價中起著重要作用,賽業生物擁有數千種自主研發的基因編輯小鼠品系,可提供包括Atp13a2、Gria3和Gldc等在內的多種基因敲除或條件性敲除罕見病研究小鼠模型。同時也可根據您的科研需求進行專業化的定制服務,加速您的課題研究。