溶酶體酸性脂肪酶缺乏癥(LAL-D)小鼠模型的介紹與應用
溶酶體酸性脂肪酶缺乏癥(lysosomal acid lipase dificiency,LAL-D)是一種罕見的、慢性的、逐漸加重的常染色體隱性遺傳性疾病。
LIPA基因會出現錯義突變、無義突變、插入/缺失、剪接位點突變、復雜重組等,導致溶酶體酸性脂肪酶(lysosomal acid lipase,LAL)活性缺失或嚴重不足(<1%的正常活性)。LAL功能缺失或降低,使溶酶體降解低密度脂蛋白、膽固醇酯和三酰甘油的功能喪失或降低,膽固醇酯和三酰甘油在組織細胞內貯積,血漿膽固醇水平升高,進而導致內臟器官出現黃色瘤樣變化。
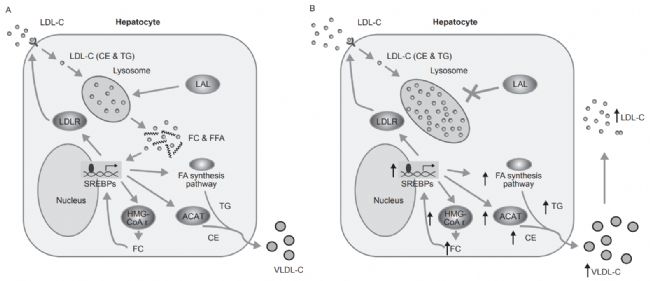
圖1. 健康個體和LAL-D患者的細胞膽固醇穩態的示意圖[1]。
根據發病年齡和臨床表現不同,分為嬰兒期起病的沃爾曼病(Wolman disease,WD)和兒童及成人期起病的膽固醇酯貯積病(cholesterol ester storage disease,CESD)。目前針對溶酶體酸性脂肪酶缺乏癥的治療方法主要包括病因治療和對癥治療。Sebelipase alfa是首個獲批的酶替代治療藥物,能改善血脂異常、肝功能、胃腸道癥狀,并延長生存期。
病因治療:造血干細胞移植和酶替代治療。
對癥支持治療:低脂飲食、胃腸外營養、糖皮質激素和鹽皮質激素替代等。
南模生物LAL-D模型
南模生物長期助力罕見病基因治療研究,構建了Lipa-KO溶酶體酸性脂肪酶缺乏癥模型,助力溶酶體酸性脂肪酶缺乏癥相關機制研究需求,為相關藥物的藥效評估和安全性評價提供了強有力的工具。
/ Lipa-KO(NM-KO-201298)/
部分驗證數據:
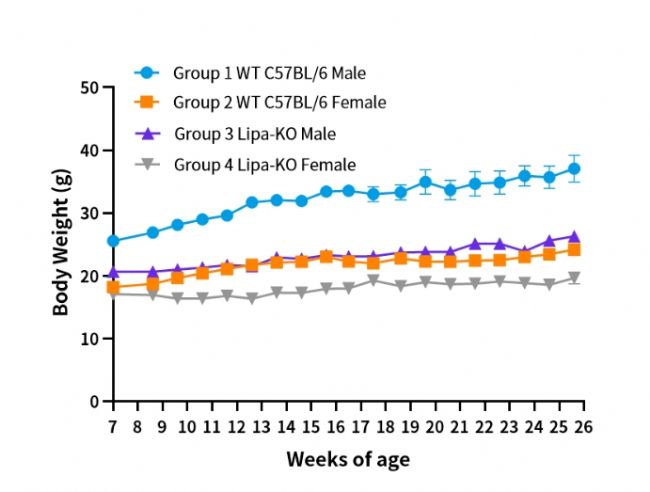
圖1. Lipa-KO小鼠體重變化。
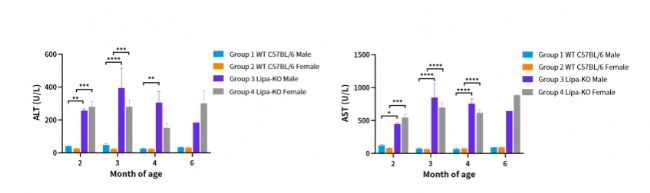
圖2. 不同月齡Lipa-KO小鼠血清中丙氨酸氨基轉移酶(alanine aminotransferase,ALT)和谷草轉氨酶(aspartate aminotransferase,AST)含量。
(2~4month: n=3~6; 6month: n=1~3)
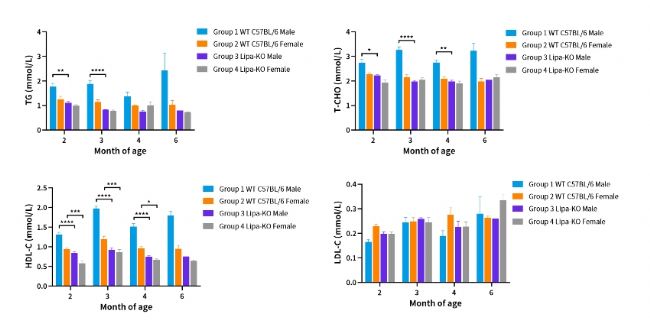
圖3. 不同月齡ipa-KO小鼠血脂分析。
(2~4month: n=3~6; 6month: n=1~3)
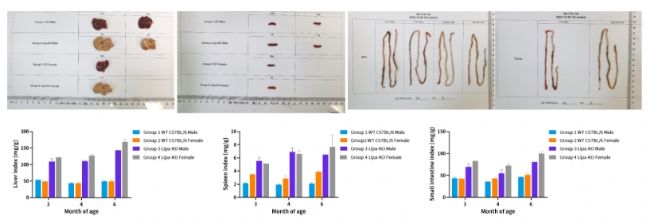
圖4. 肝臟中總膽固醇含量。(n=1~3)
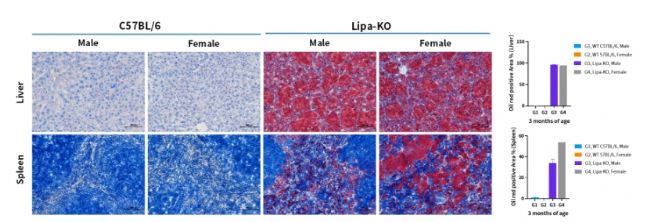
圖5. 3月齡Lipa-KO小鼠肝臟和脾臟的油紅染色代表性圖像及統計分析。
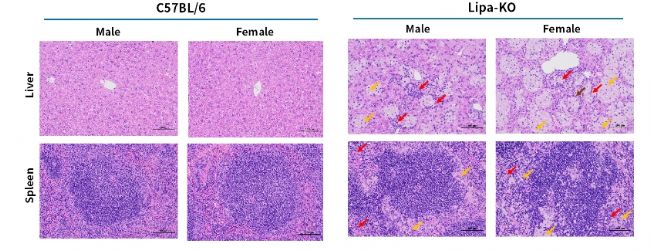
圖6. 3月齡Lipa-KO小鼠肝臟和脾臟的H&E染色代表性圖片。
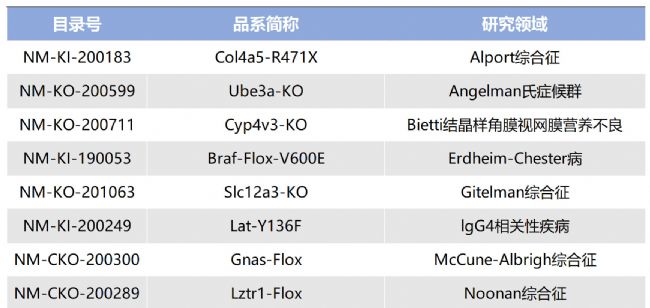
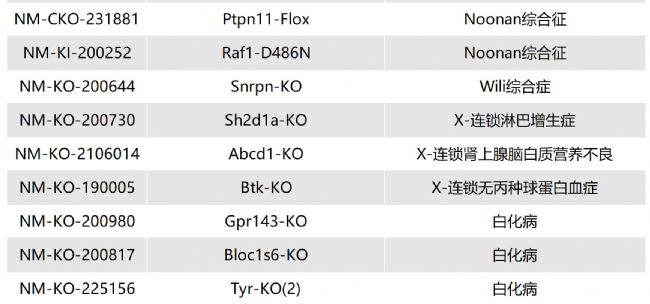
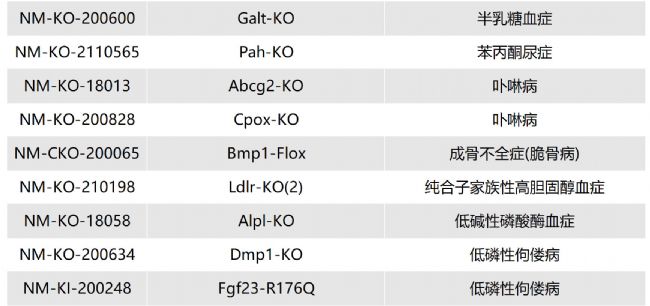
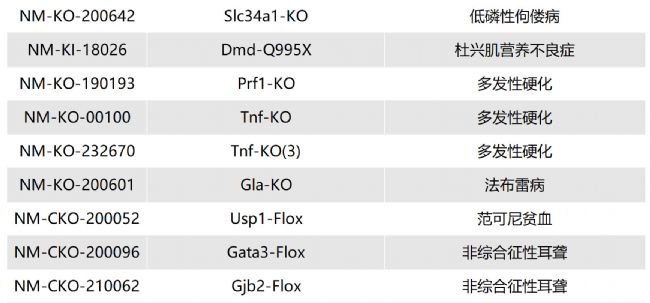
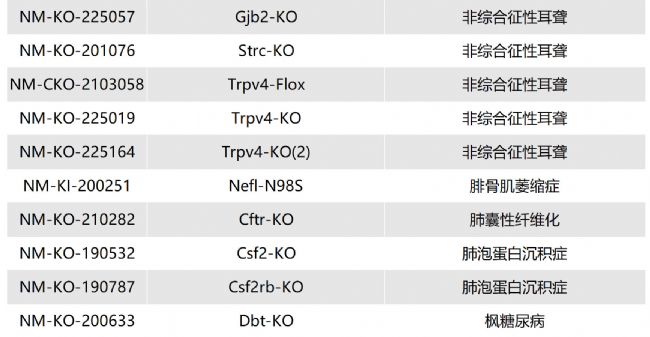
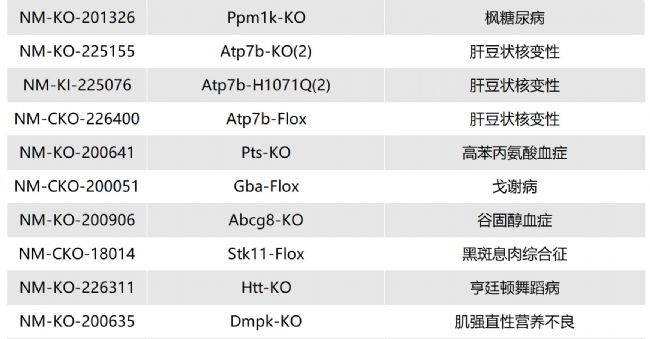
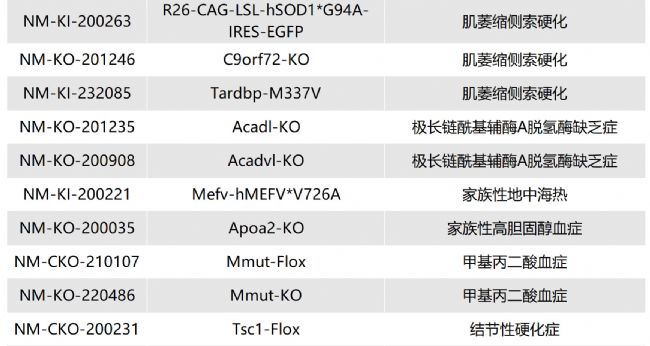
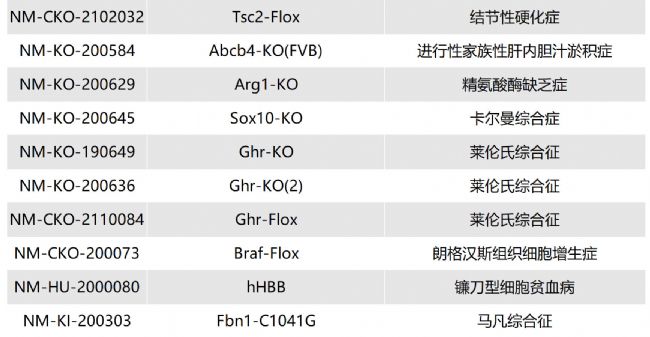
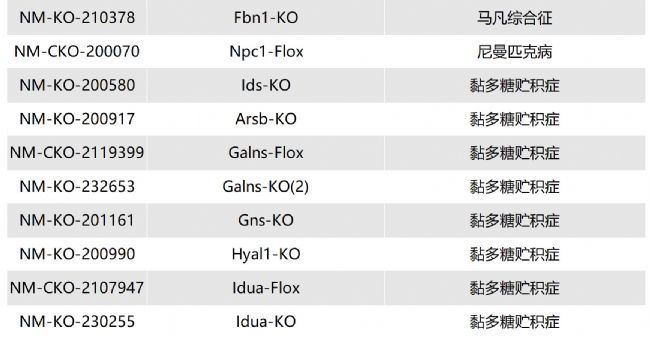
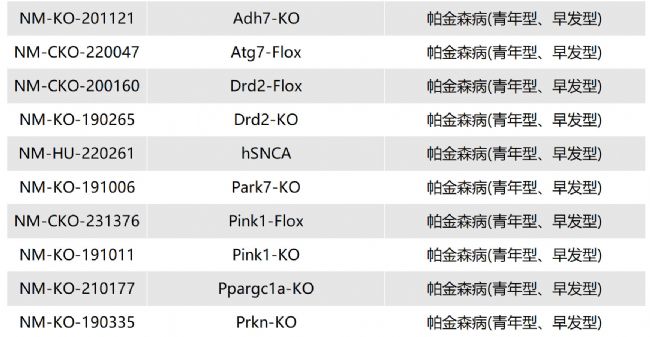
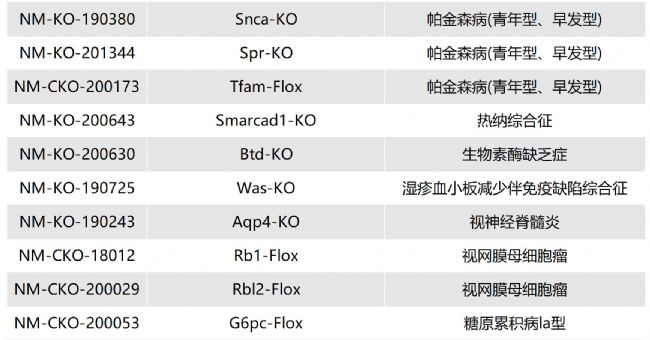
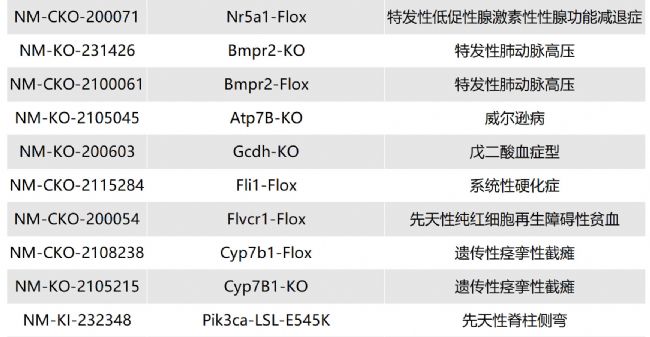
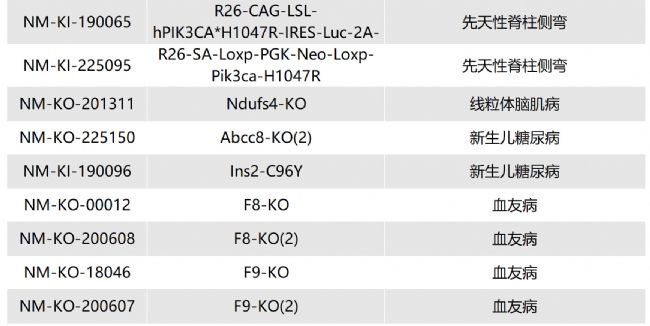
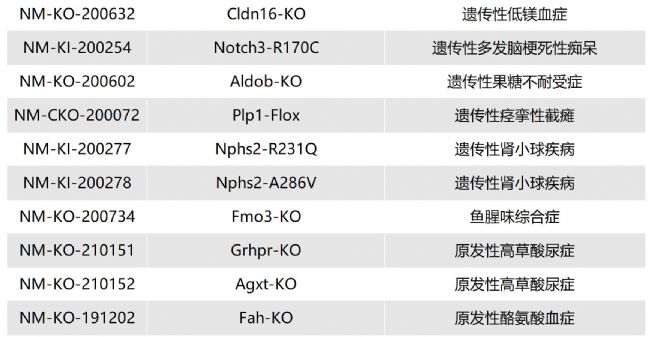
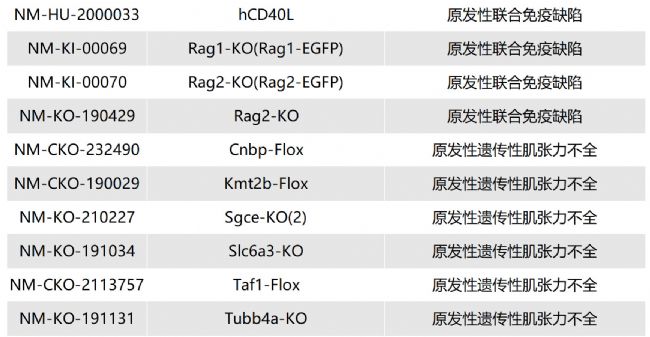

如您有相關需求,歡迎撥打400-728-0660或者于微信公眾號南模生物SMOC在線咨詢,或致信marketing@modelorg.com,我們的專業團隊將竭誠為您服務!
Reference:
[1] Reiner Ž, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiency--an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235(1):21-30. doi:10.1016/j.atherosclerosis.2014.04.003
關于我們
上海南方模式生物科技股份有限公司(Shanghai Model Organisms Center, Inc.,簡稱"南模生物"),成立于2000年9月,是一家上交所科創板上市高科技生物公司(股票代碼:688265),始終以編輯基因、解碼生命為己任,專注于模式生物領域,打造了以基因修飾動物模型研發為核心,涵蓋多物種模型構建、飼養繁育、表型分析、藥物臨床前評價等多個技術平臺,致力于為全球高校、科研院所、制藥企業等客戶提供全方位、一體化的基因修飾動物模型產品解決方案。