
AAV-RGDLRVSĻÖ÷ĶÃ}ČƤŧ°ûĩÄŪDȞЧÂĘČįēÎŖŋ
2025Äę6ÔÂ3ČÕŖŦŋÕÜÜát´ķWģųĩAátW˛ŋÉúĀíÅc˛ĄĀíÉúĀíŊĖŅĐĘŌÂēĪÎ÷°˛Ŋģͨ´ķWFę ÔÚCell MetabolismŖ¨IF=27.7ŖŠÔÚž°ląíî}é“O-GlcNAcylation-mediated endothelial metabolic memory contributes to cardiac damage via small extracellular vesicles”ĩÄŅĐžŋÕÎÄŖŦąžŅĐžŋŊŌĘžÁËŌģˇNͨß^O-GlcNAõŖģ¯ŦFČƤ´úÖxĶĩÄĐÂCÖÆŖŦ˛ĸ´_ļ¨ÁËĖĮÄō˛ĄECŅÜÉúĩÄĐĄŧ°ûÍâÄŌÅŨŖ¨sEVsŖŠĩÄĶĖØÕ÷ū§ÖÂĐÄÅKpûĄŖ


ŅĐžŋąŗž°
ĖĮÄō˛ĄŖ¨DMŖŠĩÄģŧ˛ĄÂĘī@ÖøÔöŧĶŖŦŌŅŗÉéŌģÖ÷ŌĒĩÄČĢĮōšĢš˛ĐlÉúî}ĄŖąËųÖÜÖĒŖŦĖĮÄō˛Ąī@ÖøÔöŧĶÁËĐÄÁĻËĨŊßĩÄīLëUŖŦÅcČąŅĒĐÔĐÄÅK˛ĄģōÆäËû˛ĸ°l°YĩÄ´æÔÚoęPŖģÁíŌģˇŊÃæŖŦÅR´˛×CūŌ˛ąíÃ÷ŖŦÔÚĐÄÁĻËĨŊßģŧÕßÖĐŖŦĖĮÄō˛ĄĘĮ˛ģÁŧîAēķĩÄęPæIQļ¨ŌōËØĄŖąMšÜÖίēķŅĒĖĮģÖÍÕũŗŖŖŦĩĢ¸ßŅĒĖĮŽaÉúĩÄßmĒ˛ģÁŧĖØÕ÷ČÔČģ´æÔÚĄŖūÍÆyŖŦŅĒšÜĪĩŊyÖĐÔįÆÚĩĸßŅĒĖĮŋÉÄÜūąģČƤŧ°ûŖ¨ECsŖŠĶץŖŦÄļø§ÖÂëSēķĩÄĐÄŅĒšÜ˛ĸ°l°YĄŖß@ˇNŦFĪķąģˇQ铸ßŅĒĖĮĶ”ģō“´úÖxĶ”ŖŦÖ÷ŌĒĶÃĶÚÃčĘö¸ßŅĒĖĮĻŅĒšÜ˛ĸ°l°YĩÄéLÆÚØÃæĶ°íĄŖČģļøŖŦ´úÖxĶÔÚĖĮÄō˛ĄĐÄŧĄ˛ĄģōĐÄÁĻËĨŊß°lÕšÖĐĩÄ×÷ĶÃÔÚēÜ´ķŗĖļČÉĪČÔ˛ģĮåŗūĄŖ
ŅĐžŋŊYšûˇÖĪí
1Ąĸ´úÖxĶŽaÉúĩÄsEV miR-15-16ÉĪÕ{FoxO1ĐÅĖ÷§
×÷Õßͨß^ķwČÍâōŅĐžŋ°lŦFÔÚ8ÖÜĩĸßŅĒĖĮŖ¨HGŖŠĒŧ¤ēķŖŦĖĮÄō˛ĄíÔ´ĩÄŅhĐĄŧ°ûÍâÄŌÅŨŖ¨sEVsŖŠĻĐÄŧĄŧ°ûŽaÉúŗÖĀmĐÔ˛ģĀûĶ°íŖŦ˛ĸÔÚŊĄŋĩĶÎīÖĐÕT§ĐÄÅKšĻÄÜÕĪĩKŖŦąMšÜēķĀmŅĒĖĮģÖÍÕũŗŖĄŖß@ąíÃ÷HGÕT§ĩÄŅhsEVsąíŦFŗö“Ķ”ĖØÕ÷ŖŦŋÉÄÜ´ŲĘšĖĮÄō˛ĄģŧÕßÁô˛ŋˇÖĐÄÁĻËĨŊßīLëUĄŖŋŧ]ĩŊÔÚĖĮÄō˛Ą8ÖÜrsEVsŽaÉúĩIJģÁŧĶ°íŖŦĻÄ8ÖÜĖĮÄō˛Ąē͡ĮĖĮÄō˛ĄĻÕÕŊM´ķĘķŅĒ{ÖĐĘÕŧ¯ĩÄsEVsßMĐиßͨÁŋRNAyĐōŖŦčbļ¨°lŦFmiR-15b-5pēÍmiR-16-5pĘĮ´úÖxĶŽaÉúĩÄŅhsEVsÖĐĩÄ×÷ĶÃˇÖ×ĶĄŖmiR-15b-5pēÍmiR-16-5pąģŪDäémiR-15-16ŖŦÎģĶÚÍŦŌģČžÉĢķw ^ĶōŖŦ˛ĸĮŌ¸ßļČąŖĘØŖŦŋŧ]ĩŊÉÕßĩÄ fÍŦ×÷ĶÃŖŦēķĀmĘšĶÃÆäÄŖMÎīģōŌÖÖÆŠĖŊžŋÁËmiR-15-16ÕT§ĐÄŧĄŧ°ûĩōÍöĩġÖ×ĶCÖÆĄŖŊYšûąíÃ÷ŖŦsEVs miR-15-16°ĐĪōFoxO1ĐÅĖͨ¡ĩÄŌģĪĩÁĐÉĪĶÎÕ{šŌō×ĶŖŦ˛ĸͨß^ÉĪÕ{ĐÄŧĄŧ°ûÖĐĩÄFoxO1°l]´ŲĩōÍö×÷ĶÃŖŦßMļø§ÖÂĐÄÅKšĻÄÜÕĪĩKĄŖ
×÷ÕßßMŌģ˛ŊŅĐžŋ°lŦF´úÖxĶÕT§ĶÃ}ČƤŧ°ûˇÖÃÚsEV miR-15-16ŖŦéÁËĖŊË÷ČƤmiR-15-16ÔÚķwČ´úÖxĶÕT§ĩÄĐÄÅKšĻÄÜÕĪĩKÖĐĩÄ×÷ĶÃŖŦ×÷ÕßĀûĶÃAAV9Ŋé§miR-15-16ēŖždßfËÍŊoĐĄĘķŖŦŦFÁËECĖØŽĐÔĩÄmiR-15-16ĮÃĩÍĄŖÅcNCēŖždŊMĪāąČŖŦECĖØŽĐÔmiR-15-16ēŖždŊMŅĒ{sEV miR-15-16ĩÄąíß_ī@ÖøŊĩĩÍŖŦąíÃ÷ĶÃ}ECĘĮ´úÖxĶÕT§ĩÄŅĒ{sEV miR-15-16ĩÄÖ÷ŌĒíÔ´ĄŖ´ËÍâŖŦÔÚDMģōDM+InsŖ¨´úÖxĶĶÎīÄŖĐÍŖŠŊMÖĐŖŦECĖØŽĐÔmiR-15-16ĮÃĩÍŊĩĩÍÁËĐÄŧĄmiR-15-16ĩÄąíß_ŖŦ¸ÄÉÆÁËĐÄšĻÄÜŖŦ˛ĸŌÖÖÆÁËĐÄŧĄŧ°ûĩōÍöēÍFoxO1ĩÄąíß_ĄŖß@ĐŠĩūąíÃ÷ŖŦČƤŧ°ûˇÖÃÚĩÄsEV miR-15-16 ĸÅcÁË´úÖxĶÕT§ĩÄĐÄÅKpûēÍšĻÄÜÕĪĩKĄŖ
×÷ÕßßMŌģ˛ŊĖŊžŋÁË´úÖxĶ´ŲßMECsˇÖÃÚsEV miR-15-16ĩÄCÖÆŖŦ°lŦFHGĻmiR-15-16ĩÄ×÷ĶÃŋÉÄÜͨß^ÕT§ĩ°°×Ų|O-GlcNAcĖĮģųģ¯Ŋ駥ŖĖĮÄō˛ĄĶÃ}ECsÖĐO-GlcNAcĖĮģųģ¯ĄĸCaMK2aģîĐÔĄĸCaMK2a-Stat1ŊYēĪŌÔŧ°Stat1ÔÚSer727ÎģücĩÄÁ×Ëáģ¯žųī@ÖøÉũ¸ßŖŦŧ´ĘšēķÆÚͨß^ŌČuËØÖ˛ČëŦFŅĒĖĮÕũŗŖģ¯ŖŦß@ĐŠÖ¸ËČÔī@ÖøÉũ¸ßĄŖĀûĶÃĪŲ˛ĄļžÔÚECsÖĐß^ąíß_Stat1ŖŦō°lŦFStat1ŋÉŌÔÅcpri-miR-15-16ĸĶ×ĶŊYēĪŖŦŌÔÔöECÖĐsEV miR-15-16ĩġÖÃÚĄŖëSēķŖŦ×÷Õßͨß^ŌÖÖÆCaMK2a/Stat1ĖŊžŋÁËČƤŧ°ûCaMK2a-Stat1ĐÅĖ÷§ÔÚ´úÖxĶÕT§ĩÄsEV miR-15-16ˇÖÃÚÉĪÕ{ÖĐĩÄ×÷ĶÃŖŦŊYšûąíÃ÷ČƤŧ°ûCaMK2a-Stat1ĐÅĖ÷§Ŋ駴úÖxĶÕT§ĩÄŅĒ{sEV miR-15-16ÉĪÕ{ŖŦ˛ĸÕT§ĐÄÅKšĻÄÜÕĪĩKĄŖ
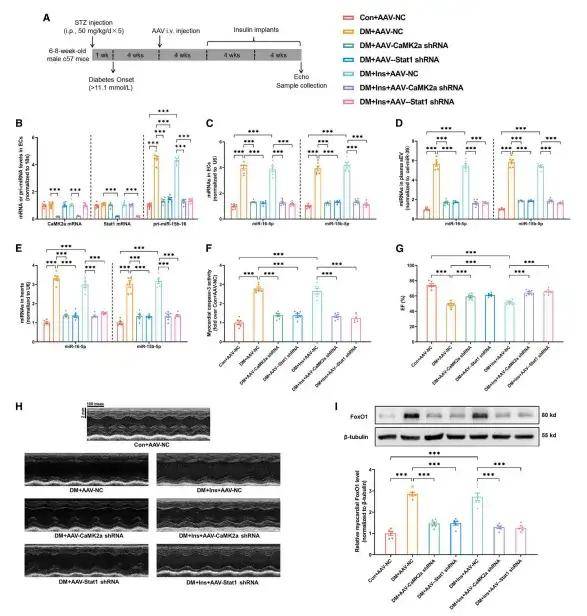
4ĄĸÔįÆÚŌČuËØÖίģōO-GlcNAcĐŪīŌÖÖÆŋÉ×čā¸ßŅĒĖĮÕT§ĩÄĶĐÅĖ˛ĸîAˇĀĖĮÄō˛ĄĐÔĐÄŧĄpû
Į°ÆÚš¤×÷čbļ¨Thr306éCaMK2aĩÄÔÚGlcNAcĐŪīÎģücŖŦĻÔÎģücßMĐĐÍģ×ÔōĄŖĀûĶÃĪŲ˛ĄļžÔÚECÖĐąíß_ÍâÔ´Ō°ÉúĐÍģōT306AÍģ×ķwCaMK2a-FlagŖŦ×CÃ÷ÁËThr306ÎģücĩÄO-GlcNAcĐŪīĘĮCaMK2a´ŲßMStat1Á×Ëáģ¯ŧ°ēķĀmsEV miR-15-16ˇÖÃÚËųąØĐčĩÄĄŖßMŌģ˛ŊōąíÃ÷O-GlcNAcĐŪīĩÄCaMK2aÔÚķwČžßĶĐēܸßĩġļ¨ĐÔĄŖCaMK2aÕ{špri-miR-15/16ĩÄąíß_ŖŦ˛ĸͨß^Á×Ëáģ¯Stat1Ķ°íĐÄŧĄŧ°ûĄŖąMšÜĖĮÄō˛Ą°l˛Ą8ÖÜēķé_ĘŧĩÄÍíÆÚŌČuËØÖίoˇ¨pŨp´úÖxĶē͡ĀÖšĐÄÅKšĻÄÜÕĪĩKĩÄ°lÕšŖŦĩĢĘĮĖŊžŋĖĮÄō˛Ą°l˛ĄŗõÆÚÔįÆÚŌČuËØÖίģōO-GlcNAcĐŪīŌÖÖÆŠŖ¨OSMIŖŦ1 mg/kg/ĖėŖŠĘĮˇņÄÜĶĐЧ×čāČƤĶĐÅĖēÍĐÄÅKpûžßĶĐÖØŌĒŌâÁxĄŖŊYšûąíÃ÷ŖŦĩ°°×Ų|O-GlcNAģ¯ÖąŊĶŊé§ĖĮÄō˛ĄÕT§ĩÄĶĐēĻ×ģ¯ŖŦļøÔįÆÚŅĒĖĮŋØÖÆÄÜōŌÖÖÆ´úÖxĶĐÅĖ÷§˛ĸîAˇĀĖĮÄō˛ĄÕT§ĩÄpûĄŖ×÷ÕßßMŌģ˛Ŋō×CÁËōŊYšûĩÄŪDģ¯ßmĶÃĐÔŖŦÄĖĮÄō˛ĄķwēÍŊĄŋĩÖžÔ¸ÕßÖĐĖáČĄÁËŅĒ{sEVsŖŦ˛ĸͨß^ļāÔĒˇÖÎö°lŦFŅĒ{sEV miR-16-5pēÍmiR-15b-5pÔÚîAˇĀĖĮÄō˛ĄģŧÕßĐÄÅKšĻÄÜÕĪĩKˇŊÃæžßĶĐŪDģ¯ÁĻĄŖ
ŊYÕ
ąžŅĐžŋąíÃ÷ĖĮÄō˛ĄģŧÕßÖίēķąMšÜŅĒĖĮģÖÍÕũŗŖŖŦĩĢ´úÖxĶŅÜÉúĩÄŅhsEV miR-15-16ĻĐÄŧĄŧ°ûŽaÉúÁËÉîßhļøŗÖĀmĩÄĶĐēĻĶ°íĄŖ¸ßĖĮÕT§ĩÄCaMK2a/O-GlcNAģ¯ĩÄÕũˇ´đģØ¡ŧ´ĘšÔÚGLUÕũŗŖģ¯ēķŌ˛ÄÜžSŗÖČƤCaMK2a-Stat1ͨ¡ĩÄŧ¤ģîŖŦÔͨ¡Ŋ駴úÖxĶ˛ĸ´ŲßMsEV miR-15-16ĩÄŗÖĀmáˇÅĄŖ°ĐĪōČƤO-GlcNAc-CaMK2a-Stat1ͨ¡ģōsEV miR-15-16éĪûŗũĖĮÄō˛Ą´úÖxĶŧ°ÆäĪāęP˛ĸ°l°YĖᚊÁËŌģˇNĶĐĮ°ž°ĩÄÖίˇŊˇ¨ĄŖ
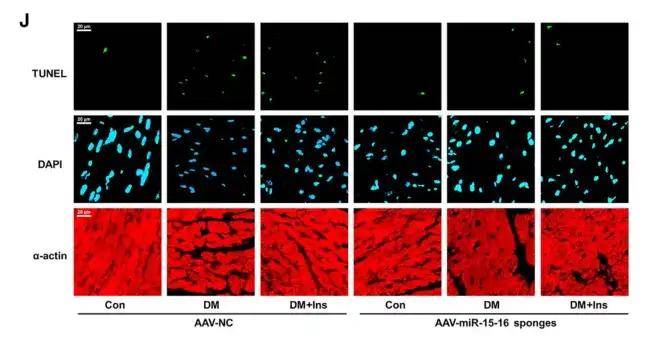
AAV¸ĐČžēķTUNELēÍDAPIČžÉĢĩÄ´úąíĐÔDĪņ
ĐĄĘķîiĶÃ}ÖĐCD31Ŗ¨ČƤŧ°ûËÖžÎīŖŠÅcCaMK2a-Flagš˛ÃâŌßÉšâČžÉĢĩÄ´úąíĐÔDĪņ
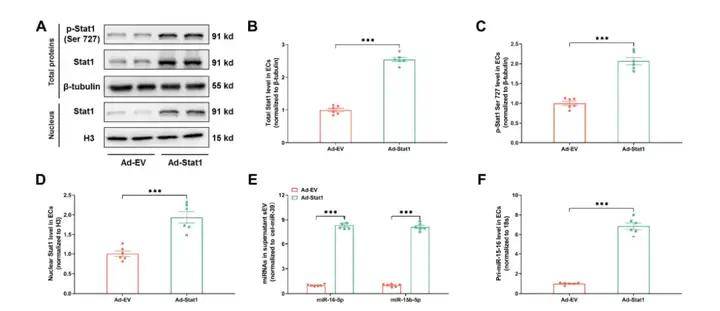
Ad-Stat1ąíß_ēķ Stat1ŧ°miR-16-5p ĩČĪāęPģųŌōĩÄËŽÆŊzy
ŅĐžŋąŗž°
ĖĮÄō˛ĄŖ¨DMŖŠĩÄģŧ˛ĄÂĘī@ÖøÔöŧĶŖŦŌŅŗÉéŌģÖ÷ŌĒĩÄČĢĮōšĢš˛ĐlÉúî}ĄŖąËųÖÜÖĒŖŦĖĮÄō˛Ąī@ÖøÔöŧĶÁËĐÄÁĻËĨŊßĩÄīLëUŖŦÅcČąŅĒĐÔĐÄÅK˛ĄģōÆäËû˛ĸ°l°YĩÄ´æÔÚoęPŖģÁíŌģˇŊÃæŖŦÅR´˛×CūŌ˛ąíÃ÷ŖŦÔÚĐÄÁĻËĨŊßģŧÕßÖĐŖŦĖĮÄō˛ĄĘĮ˛ģÁŧîAēķĩÄęPæIQļ¨ŌōËØĄŖąMšÜÖίēķŅĒĖĮģÖÍÕũŗŖŖŦĩĢ¸ßŅĒĖĮŽaÉúĩÄßmĒ˛ģÁŧĖØÕ÷ČÔČģ´æÔÚĄŖūÍÆyŖŦŅĒšÜĪĩŊyÖĐÔįÆÚĩĸßŅĒĖĮŋÉÄÜūąģČƤŧ°ûŖ¨ECsŖŠĶץŖŦÄļø§ÖÂëSēķĩÄĐÄŅĒšÜ˛ĸ°l°YĄŖß@ˇNŦFĪķąģˇQ铸ßŅĒĖĮĶ”ģō“´úÖxĶ”ŖŦÖ÷ŌĒĶÃĶÚÃčĘö¸ßŅĒĖĮĻŅĒšÜ˛ĸ°l°YĩÄéLÆÚØÃæĶ°íĄŖČģļøŖŦ´úÖxĶÔÚĖĮÄō˛ĄĐÄŧĄ˛ĄģōĐÄÁĻËĨŊß°lÕšÖĐĩÄ×÷ĶÃÔÚēÜ´ķŗĖļČÉĪČÔ˛ģĮåŗūĄŖ
ŅĐžŋŊYšûˇÖĪí
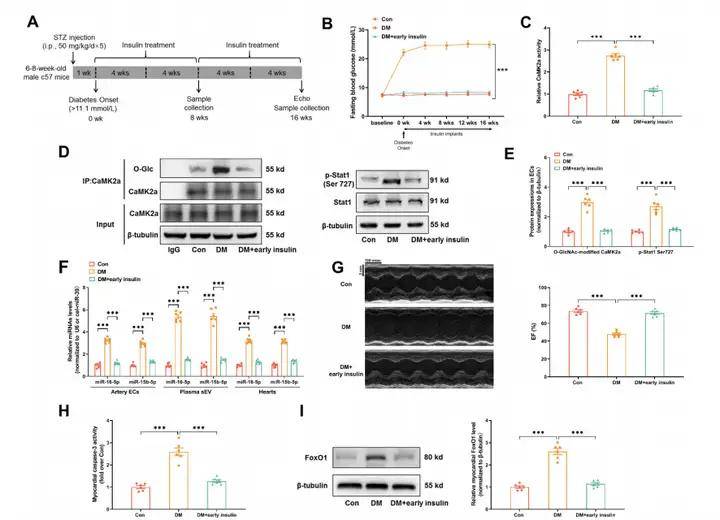
1Ąĸ´úÖxĶŽaÉúĩÄsEV miR-15-16ÉĪÕ{FoxO1ĐÅĖ÷§
×÷Õßͨß^ķwČÍâōŅĐžŋ°lŦFÔÚ8ÖÜĩĸßŅĒĖĮŖ¨HGŖŠĒŧ¤ēķŖŦĖĮÄō˛ĄíÔ´ĩÄŅhĐĄŧ°ûÍâÄŌÅŨŖ¨sEVsŖŠĻĐÄŧĄŧ°ûŽaÉúŗÖĀmĐÔ˛ģĀûĶ°íŖŦ˛ĸÔÚŊĄŋĩĶÎīÖĐÕT§ĐÄÅKšĻÄÜÕĪĩKŖŦąMšÜēķĀmŅĒĖĮģÖÍÕũŗŖĄŖß@ąíÃ÷HGÕT§ĩÄŅhsEVsąíŦFŗö“Ķ”ĖØÕ÷ŖŦŋÉÄÜ´ŲĘšĖĮÄō˛ĄģŧÕßÁô˛ŋˇÖĐÄÁĻËĨŊßīLëUĄŖŋŧ]ĩŊÔÚĖĮÄō˛Ą8ÖÜrsEVsŽaÉúĩIJģÁŧĶ°íŖŦĻÄ8ÖÜĖĮÄō˛Ąē͡ĮĖĮÄō˛ĄĻÕÕŊM´ķĘķŅĒ{ÖĐĘÕŧ¯ĩÄsEVsßMĐиßͨÁŋRNAyĐōŖŦčbļ¨°lŦFmiR-15b-5pēÍmiR-16-5pĘĮ´úÖxĶŽaÉúĩÄŅhsEVsÖĐĩÄ×÷ĶÃˇÖ×ĶĄŖmiR-15b-5pēÍmiR-16-5pąģŪDäémiR-15-16ŖŦÎģĶÚÍŦŌģČžÉĢķw ^ĶōŖŦ˛ĸĮŌ¸ßļČąŖĘØŖŦŋŧ]ĩŊÉÕßĩÄ fÍŦ×÷ĶÃŖŦēķĀmĘšĶÃÆäÄŖMÎīģōŌÖÖÆŠĖŊžŋÁËmiR-15-16ÕT§ĐÄŧĄŧ°ûĩōÍöĩġÖ×ĶCÖÆĄŖŊYšûąíÃ÷ŖŦsEVs miR-15-16°ĐĪōFoxO1ĐÅĖͨ¡ĩÄŌģĪĩÁĐÉĪĶÎÕ{šŌō×ĶŖŦ˛ĸͨß^ÉĪÕ{ĐÄŧĄŧ°ûÖĐĩÄFoxO1°l]´ŲĩōÍö×÷ĶÃŖŦßMļø§ÖÂĐÄÅKšĻÄÜÕĪĩKĄŖ
D1. ´úÖxĶŽaÉúĩÄsEV miR-15-16ÉĪÕ{FoxO1ĐÅĖ÷§
2ĄĸŌÖÖÆČƤŧ°ûmiR-15-16ŋÉpŨp´úÖxĶÕT§ĩÄĐÄÅKpûēÍšĻÄÜÕĪĩK×÷ÕßßMŌģ˛ŊŅĐžŋ°lŦF´úÖxĶÕT§ĶÃ}ČƤŧ°ûˇÖÃÚsEV miR-15-16ŖŦéÁËĖŊË÷ČƤmiR-15-16ÔÚķwČ´úÖxĶÕT§ĩÄĐÄÅKšĻÄÜÕĪĩKÖĐĩÄ×÷ĶÃŖŦ×÷ÕßĀûĶÃAAV9Ŋé§miR-15-16ēŖždßfËÍŊoĐĄĘķŖŦŦFÁËECĖØŽĐÔĩÄmiR-15-16ĮÃĩÍĄŖÅcNCēŖždŊMĪāąČŖŦECĖØŽĐÔmiR-15-16ēŖždŊMŅĒ{sEV miR-15-16ĩÄąíß_ī@ÖøŊĩĩÍŖŦąíÃ÷ĶÃ}ECĘĮ´úÖxĶÕT§ĩÄŅĒ{sEV miR-15-16ĩÄÖ÷ŌĒíÔ´ĄŖ´ËÍâŖŦÔÚDMģōDM+InsŖ¨´úÖxĶĶÎīÄŖĐÍŖŠŊMÖĐŖŦECĖØŽĐÔmiR-15-16ĮÃĩÍŊĩĩÍÁËĐÄŧĄmiR-15-16ĩÄąíß_ŖŦ¸ÄÉÆÁËĐÄšĻÄÜŖŦ˛ĸŌÖÖÆÁËĐÄŧĄŧ°ûĩōÍöēÍFoxO1ĩÄąíß_ĄŖß@ĐŠĩūąíÃ÷ŖŦČƤŧ°ûˇÖÃÚĩÄsEV miR-15-16 ĸÅcÁË´úÖxĶÕT§ĩÄĐÄÅKpûēÍšĻÄÜÕĪĩKĄŖ
D2. ŌÖÖÆČƤŧ°ûmiR-15-16ŋÉpŨp´úÖxĶÕT§ĩÄĐÄÅKpûēÍšĻÄÜÕĪĩK
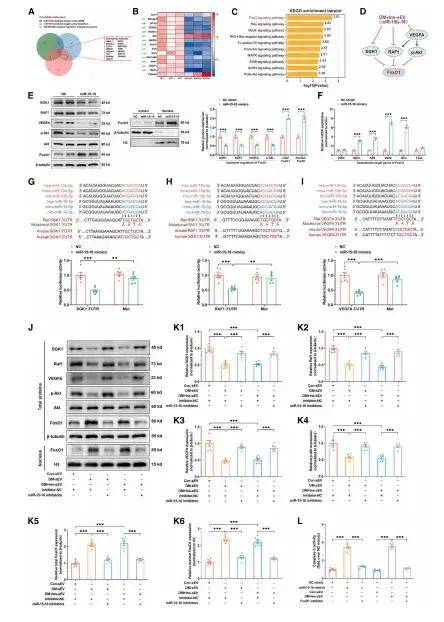
3ĄĸĮÃĩÍČƤŧ°ûCaMK2a-Stat1ĐÅĖŪD§ŌÖÖÆ´úÖxĶÕT§ĩÄsEV miR-15-16ˇÖÃÚēÍĐÄÅKšĻÄÜÕĪĩK×÷ÕßßMŌģ˛ŊĖŊžŋÁË´úÖxĶ´ŲßMECsˇÖÃÚsEV miR-15-16ĩÄCÖÆŖŦ°lŦFHGĻmiR-15-16ĩÄ×÷ĶÃŋÉÄÜͨß^ÕT§ĩ°°×Ų|O-GlcNAcĖĮģųģ¯Ŋ駥ŖĖĮÄō˛ĄĶÃ}ECsÖĐO-GlcNAcĖĮģųģ¯ĄĸCaMK2aģîĐÔĄĸCaMK2a-Stat1ŊYēĪŌÔŧ°Stat1ÔÚSer727ÎģücĩÄÁ×Ëáģ¯žųī@ÖøÉũ¸ßŖŦŧ´ĘšēķÆÚͨß^ŌČuËØÖ˛ČëŦFŅĒĖĮÕũŗŖģ¯ŖŦß@ĐŠÖ¸ËČÔī@ÖøÉũ¸ßĄŖĀûĶÃĪŲ˛ĄļžÔÚECsÖĐß^ąíß_Stat1ŖŦō°lŦFStat1ŋÉŌÔÅcpri-miR-15-16ĸĶ×ĶŊYēĪŖŦŌÔÔöECÖĐsEV miR-15-16ĩġÖÃÚĄŖëSēķŖŦ×÷Õßͨß^ŌÖÖÆCaMK2a/Stat1ĖŊžŋÁËČƤŧ°ûCaMK2a-Stat1ĐÅĖ÷§ÔÚ´úÖxĶÕT§ĩÄsEV miR-15-16ˇÖÃÚÉĪÕ{ÖĐĩÄ×÷ĶÃŖŦŊYšûąíÃ÷ČƤŧ°ûCaMK2a-Stat1ĐÅĖ÷§Ŋ駴úÖxĶÕT§ĩÄŅĒ{sEV miR-15-16ÉĪÕ{ŖŦ˛ĸÕT§ĐÄÅKšĻÄÜÕĪĩKĄŖ
D3. ĮÃĩÍČƤŧ°ûCaMK2a-Stat1ĐÅĖŪD§ŌÖÖÆ´úÖxĶÕT§ĩÄsEV miR-15-16ˇÖÃÚēÍĐÄÅKšĻÄÜÕĪĩK
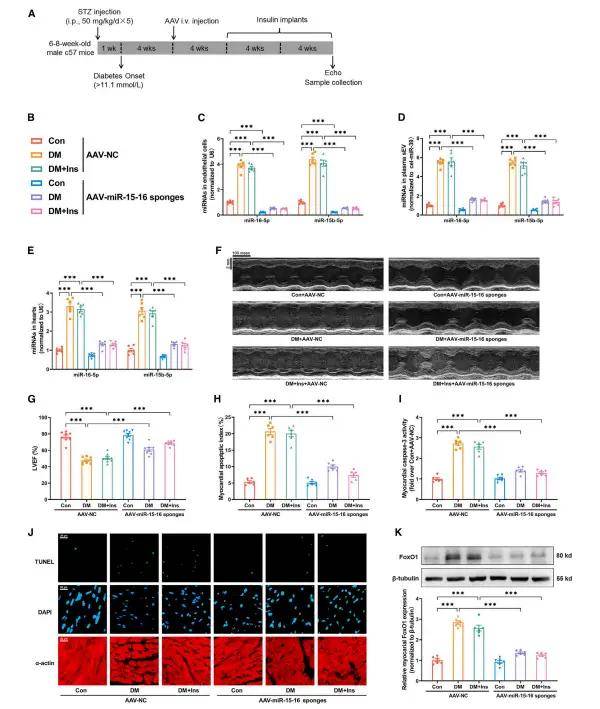
4ĄĸÔįÆÚŌČuËØÖίģōO-GlcNAcĐŪīŌÖÖÆŋÉ×čā¸ßŅĒĖĮÕT§ĩÄĶĐÅĖ˛ĸîAˇĀĖĮÄō˛ĄĐÔĐÄŧĄpû
Į°ÆÚš¤×÷čbļ¨Thr306éCaMK2aĩÄÔÚGlcNAcĐŪīÎģücŖŦĻÔÎģücßMĐĐÍģ×ÔōĄŖĀûĶÃĪŲ˛ĄļžÔÚECÖĐąíß_ÍâÔ´Ō°ÉúĐÍģōT306AÍģ×ķwCaMK2a-FlagŖŦ×CÃ÷ÁËThr306ÎģücĩÄO-GlcNAcĐŪīĘĮCaMK2a´ŲßMStat1Á×Ëáģ¯ŧ°ēķĀmsEV miR-15-16ˇÖÃÚËųąØĐčĩÄĄŖßMŌģ˛ŊōąíÃ÷O-GlcNAcĐŪīĩÄCaMK2aÔÚķwČžßĶĐēܸßĩġļ¨ĐÔĄŖCaMK2aÕ{špri-miR-15/16ĩÄąíß_ŖŦ˛ĸͨß^Á×Ëáģ¯Stat1Ķ°íĐÄŧĄŧ°ûĄŖąMšÜĖĮÄō˛Ą°l˛Ą8ÖÜēķé_ĘŧĩÄÍíÆÚŌČuËØÖίoˇ¨pŨp´úÖxĶē͡ĀÖšĐÄÅKšĻÄÜÕĪĩKĩÄ°lÕšŖŦĩĢĘĮĖŊžŋĖĮÄō˛Ą°l˛ĄŗõÆÚÔįÆÚŌČuËØÖίģōO-GlcNAcĐŪīŌÖÖÆŠŖ¨OSMIŖŦ1 mg/kg/ĖėŖŠĘĮˇņÄÜĶĐЧ×čāČƤĶĐÅĖēÍĐÄÅKpûžßĶĐÖØŌĒŌâÁxĄŖŊYšûąíÃ÷ŖŦĩ°°×Ų|O-GlcNAģ¯ÖąŊĶŊé§ĖĮÄō˛ĄÕT§ĩÄĶĐēĻ×ģ¯ŖŦļøÔįÆÚŅĒĖĮŋØÖÆÄÜōŌÖÖÆ´úÖxĶĐÅĖ÷§˛ĸîAˇĀĖĮÄō˛ĄÕT§ĩÄpûĄŖ×÷ÕßßMŌģ˛Ŋō×CÁËōŊYšûĩÄŪDģ¯ßmĶÃĐÔŖŦÄĖĮÄō˛ĄķwēÍŊĄŋĩÖžÔ¸ÕßÖĐĖáČĄÁËŅĒ{sEVsŖŦ˛ĸͨß^ļāÔĒˇÖÎö°lŦFŅĒ{sEV miR-16-5pēÍmiR-15b-5pÔÚîAˇĀĖĮÄō˛ĄģŧÕßĐÄÅKšĻÄÜÕĪĩKˇŊÃæžßĶĐŪDģ¯ÁĻĄŖ
D4. ÔįÆÚŌČuËØÖίģōO-GlcNAcĐŪīŌÖÖÆŋÉ×čā¸ßŅĒĖĮÕT§ĩÄĶĐÅĖ˛ĸîAˇĀĖĮÄō˛ĄĐÔĐÄŧĄpû
ŊYÕ
ąžŅĐžŋąíÃ÷ĖĮÄō˛ĄģŧÕßÖίēķąMšÜŅĒĖĮģÖÍÕũŗŖŖŦĩĢ´úÖxĶŅÜÉúĩÄŅhsEV miR-15-16ĻĐÄŧĄŧ°ûŽaÉúÁËÉîßhļøŗÖĀmĩÄĶĐēĻĶ°íĄŖ¸ßĖĮÕT§ĩÄCaMK2a/O-GlcNAģ¯ĩÄÕũˇ´đģØ¡ŧ´ĘšÔÚGLUÕũŗŖģ¯ēķŌ˛ÄÜžSŗÖČƤCaMK2a-Stat1ͨ¡ĩÄŧ¤ģîŖŦÔͨ¡Ŋ駴úÖxĶ˛ĸ´ŲßMsEV miR-15-16ĩÄŗÖĀmáˇÅĄŖ°ĐĪōČƤO-GlcNAc-CaMK2a-Stat1ͨ¡ģōsEV miR-15-16éĪûŗũĖĮÄō˛Ą´úÖxĶŧ°ÆäĪāęP˛ĸ°l°YĖᚊÁËŌģˇNĶĐĮ°ž°ĩÄÖίˇŊˇ¨ĄŖ

- ÖØŊMĪŲĪāęP˛Ąļž°˛ČĢáŖŋ
- SimoaŧŧĐgŊŌĘžīĘŗŌōËØÅcAD˛ĄĀíÉúÎīËÖžÎīĩÄČÔÚÂĪĩ
- ŋÕégŊMWŧŧĐgÖúÁĻ¸ßžĢļČŊâÎöˇÎĀwžSģ¯ŋÕégŅŨģ¯ÂˇŊ
- ImmPortĩūėĪÂŨdŊĖŗĖŖēShared DataÄŖKĘšĶÃˇŊˇ¨
- AAV-RGDLRVSĻÖ÷ĶÃ}ČƤŧ°ûĩÄŪDȞЧÂĘČįēÎŖŋ
- ļËÁŖøģîĐÔzyŖēČžÁĪˇ¨Åc Taqman ĖŊᡨĩÄąČŨ^ÅcδíÚ Ũ
- AAV˛Ąļž°üŅbÁ÷ŗĖŊâÎö
- ÉúÎīÖÆËŽaÆˇÖĐÄÄĐŠĶÆˇĐčŌĒ×öÖ§ÔķwzyÄØ
- ÉúÎīĐžÆŦŋÕégļāŊMWÆŊÅ_ÔŲĖíŌģÅ_CosMx SMIŗÉĪņĪĩŊy
- ÖŪĘ×Å_CellXpress.aiÔÚÉĪēŖÉúÎīĐžÆŦÍęŗÉŅbC
- SBC CosMX WTxÎŧ°ûŋÕégČĢŪDäŊMÕũĘŊé_ˇÅîAĶ
- ÉúÎīĐžÆŦÍÆŗö"ĐžŋÕŌģĖ"ˇŊ°¸ÖØËÜŋÕégļāŊMWĐ¡ļĘŊ
- IPHASEÕnĖÃé_ÕnĀ˛ŖēŧžúģØÍÍģ×ÔōŗŖŌî}ÅcŊâ´đ
- ÉúÎīĐžÆŦÅcĐžŗŦÉúÎīÂēĪÍÆŗö"ĐžŋÕŌģĖ"ļāŊMWˇŊ°¸
- ÉúÎīátËzyÅcÔŠšŠĒÉĖŌíēÍÉúÎīĪōČĢøÕĐÄŧŊäNÉĖ
- žÉĪÕnŗĖŖēØiÄcĩĀšÚ ļžÅcČËķwoÂĩÄ´úÖxļˇ


Copyright(C) 1998-2025 ÉúÎīÆ÷˛ÄžW ëÔŖē021-64166852;13621656896 E-mailŖēinfo@bio-equip.com